
Group 13 Cations...
The elements of the group 13 share a ns2np1 electron configuration of the valence shell in the ground state, so that the oxidation states of +I and +III are most common. However, the +III oxidation state is more stable for the group 13 metals, except for thallium. Thus, metastable univalent gallium and indium species – the focus of our group – are accessible most likely as a result of the “inert-pair” effect of the ns2 (n = 4, 5) lone pair orbital and d-block contraction. Nevertheless, both tend to disproportionate to MIII compounds and M0 metal. The introduction of positive charge yielding a M+ cation already very early on proved as a viable way to univalent MI compounds, for example with the preparation of the mixed valent MI[MIIIX4] (X = Cl, Br, I) salts. Yet, even though M[MX4] does contain a M+ ion that can be solvated in aromatic solvents, the coordinating [MX4]– anion often undergoes unwanted side-reactions and dis- or comproportionations take place in many cases. This prevented the development of a coordination chemistry especially of Ga+ for a long time. Only with the synthesis of perfluoroalkoxy-aluminate salts of type [M(arene)n]+[pf]– (n = 2,3; [pf]– = [Al(ORF)4]–, RF = C(CF3)3) that were first synthesized some years ago by us for Ga, and only a few months later in the group of Scheer for In, the field was advanced.
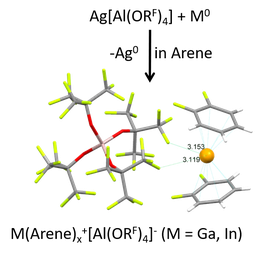

Note that the gallium p-orbitals play the main role in electron acceptance from the π-basic arenes. In contrast, the lone-pair orbital located at the gallium center remains largely unaffected by the arene-bonding. This results in a significant contraction and stabilization of the 4s2-electrons at the metal induced by the positive charge. Hence, the susceptibility of GaI towards dispropotionation is effectively inhibited. The cationic gallium arene complexes are characterized by comparatively high HOMO-LUMO-gaps > 6 eV. However, especially for the PhF-complex, the LUMO with high p-character at the gallium cation remains at low-energy, allowing for the complex to react as π-acid.
Coordination Chemistry
The M+ central ion may interact with a variety of ligands, ranging from soft (phosphines, carbenes) to hard (pyrazine, crown ethers). The coordination numbers strongly depend on the steric bulk and denticity of the ligands.

Review: Z. Anorg. Allg. Chem. online.
The nature of the frontier orbitals at dicoordinate Ga+ complexes are reminiscent to the orbital situation in carbenes or its heavier homologues (see figure).

Yet, a beautiful example on the limit of stability, is the salt [Ga(COD)2]+[pf]–, the first example for a homoleptic olefin main group metal complex. (Angew. Chem., Int. Ed. 2021, 60, 208-211.)

Quantum Theory of Atoms In Molecules (QTAIM) analysis revealed the GaI-olefine interaction to be predominantly ionic of nature and better comparable to the corresponding Na+-complex than to transition‑metal olefin‑complexes.
Cluster Chemistry
Although synthetic routes to neutral and anionic GaI and InI clusters have already been established in the early 2000s, first examples of their cationic counterparts were only synthesized in recent years. Cationic clustering may be induced by application of suitable ligands and large WCAs. Unexpectedly upon combination of the univalent indium source [In(PhF)2]+[pf]– with 1,10-phenantroline (phen) or 2,2′-bipyridine (bipy), [Inx(bipy)y]z+ cluster cations (x = 3, 4; y = 5, 6; z = 3,4) formed. (NATURE COMMUNICATIONS 2015 6:8288.)

A suggestion for the bonding interaction accounting for this structural element is included with the figure and bases on a central [(phen)In=In(phen)]2+ dication in a slight trans-bent arrangement. The p*-orbital of the In=In bond then accepts the electron density of two triplet [In(phen)2]+ moieties and produces thus the 2e3c bonds across the formal In=In double bond. Yet, the p*-orbital is only partially occupied by this interaction and thus net a very short In-In interaction results.

After a lot of synthetic efforts, we identified ligands to prepare two highly unusual examples for cationic GaI clusters. t-Butylisonitrile (CN-tBu) and 4-(dimethylamino)pyridine (DMAP) represent strong σ‑donors with good π‑donating properties and a lower tendency for redox‑non‑innocence compared to phen and bipy.

Bond Activations at Ga: In-between Cluster and Carbene Analogues...
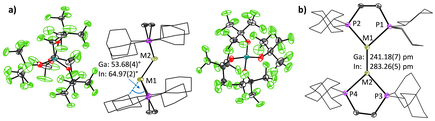
The reaction of bisdicyclohexylphosphinoethane (dcpe) and the subvalent MI sources [MI(PhF)2][pf] (M = Ga+, In+; [pf]− = [Al(ORF)4]−; RF = C(CF3)3) yielded the salts [{M(dcpe)}2][pf]2, containing the first dicationic, trans-bent digallene and diindene structures reported so far.
The non-classical MI⇆MI double bonds are surprisingly short and display a ditetrylene-like structure. The bonding situation was extensively analyzed by quantum chemical calculations, QTAIM (Quantum Theory of Atoms in Molecules) and EDA-NOCV analyses (Energy Decomposition Analysis with the combination of Natural Orbitals for Chemical Valence) and is compared to that in the isoelectronic and isostructural, but neutral digermenes and distannenes. The dissolved [{Ga(dcpe)}2]2+([pf]−)2 readily reacts with 1-hexene, cyclooctyne, diphenyldisulfide, diphenylphosphine and under mild conditions at room temperature. Angew. Chem., Int. Ed. Engl. 2023, 62, e202311648.

Ga+-Toolbox for Bond Activation: The preparation of reactive low-valent main-group complexes capable of bond activation commonly demands for multi-step processes, limiting the options of the applied systems for electronic or steric fine-tuning. Hence, here we present a systematic study on the one-pot synthesis of highly reactive Ga(I) complex cations for the activation of strong bonds that works analogously to the in-situ preparation of active transition-metal catalyst systems.